Basics

Healthcare: Innovation and Discovery
Health Care
Published on May 30, 2019
Overview
The Centers for Disease Control and Prevention’s most recent data on prescription drug use in the United States estimates that nearly fifty percent of Americans use at least one prescription drug. From the very first treatments developed, to the first vaccine to prevent smallpox in the 1790s, to the first biosimilar approved by the Food and Drug Administration in 2015, Americans have relied on this innovation to prevent diseases, cure illnesses, and alleviate pain.
It is difficult to predict the sort of cures and treatments that will be available in the future, but reminders of the progress we’ve made are everywhere. For instance, as late as the early 1950s, polio was still paralyzing tens of thousands of children a year, yet by 1979, vaccination had effectively eradicated the disease and no cases have originated in the United States since. Additionally, a diagnosis of HIV/AIDS thirty years ago carried an average life expectancy of one year, yet those diagnosed today can expect a near-normal lifespan with the proper treatment. The pace of discovery and development of treatments for these diseases, at times when scientists operated without the modern technology they employ now, should give us hope for what will be available in the future.
In this Basic, we break down the process of pharmaceutical innovation and how drugs make it to the medicine cabinet.
Research and Development
It seems we hear every week about new compounds being discovered or clinical trials yielding positive results in patients, but these treatments have usually been in the works for several years. According to the Medicare Payment Advisory Commission (MedPAC), an independent Congressional agency established to advise Congress on issues affecting Medicare, it can take roughly ten years to develop a brand new therapy. Generic versions of brand-name medications can typically take three to five years to develop. Biosimilars, like biological products, are more complex to develop and can take almost as long to produce as new therapies at roughly eight to ten years.
As medicine becomes more specialized and complex with the rise of biologic and biosimilar production, the cost of creating therapies can reach hundreds of millions to billions of dollars. Research estimates vary from about $650 million to $2.6 billion per drug based on the itemizations included, such as the research and development costs of both drugs that make it across the finish line and those that fail in clinical trials, as well as the cost of capital, or what investors give up when they commit resources to certain projects over others.
Because generics and biosimilars are modeled after existing products, they can usually be developed and brought to market for less. Generics typically cost $1 million to $5 million to develop and biosimilars about $100 million to $300 million.
The federal government plays an important role in the development of novel treatments from research to approval to patenting. In the research phase, it supports biomedical research by conducting studies, usually through the National Institutes of Health (NIH), as well as through grants and awards to research institutions around the country.
Despite the Trump Administration’s efforts to cut funding to the NIH, Congressional leaders have consistently fought for an adequate stream of funds to allow NIH to continue their research. One study published in the New England Journal of Medicine (NEJM) found that, of the 1,541 new-drug applications approved by the FDA from 1990 to 2007, 143 resulted from public-sector research institutions, or 9.3 percent.
In the research process, the NIH typically focuses on the biological targets of the drugs (basic research) instead of the drugs themselves (applied research). One study published in the Proceedings of the National Academy of Sciences (PNAS) found that NIH studies contributed in some way to all of the 210 new drugs approved by the FDA between 2010 and 2016. In ninety percent of those, the NIH contributed the basic research to complement the manufacturing industry’s applied research.
This relationship between the NIH and manufacturers is a matter of tens of billions of dollars per year and is necessary to sustain the U.S. innovation pipeline for cures.
FDA Approval
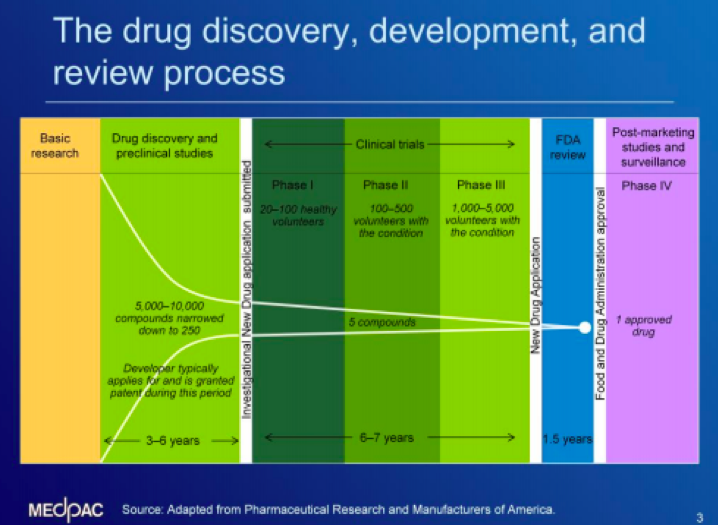
According to MedPAC, relatively few compounds make it all the way through to FDA approval. To get a new drug to market, manufacturers can begin preclinical studies with thousands of proposed drug compounds before narrowing the pool over three to six years. Preclinical research is meant to answer basic questions about safety by conducting laboratory and animal testing. From there, a manufacturer proceeds to clinical trials where a small number of compounds are tested on people through three phases to make sure the drugs are safe and effective. The studies generally include subjects with and without the condition for which the drug is being developed and can take about six to seven years to complete. Finally, a new drug application is submitted to the FDA, which conducts a thorough review of the safety and efficacy before approval, a process that can take 18 months.
More information on the drug development process can be found HERE. The chart below from MedPAC outlines the process to bring a new drug from research to approval.
To get a generic medicine approved, companies need to demonstrate “bioequivalence” which proves the medicines work in the same way and provide the same clinical benefit. Biological products, or biologics, are generally large, complex drugs manufactured using living organisms such as plant or animal cells. Because of the complicated manufacturing process required, small differences between batches of the same biologic are normal and expected, according to the FDA.
Likewise, a biosimilar cannot be exactly the same as the biologic it is modeled after. Instead, biosimilar manufacturers have to demonstrate their product has “no clinically meaningful differences” from the reference product, and that it is as safe and effective as the original biologic. All FDA-approved biological products, including biosimilars, undergo a rigorous evaluation.
Biosimilar FDA approval applications must include several of the same data as any other biologic, including studies proving their product’s similarity to a reference product, animal studies to ensure the product is not toxic, and clinical trial data to demonstrate safety. Biosimilar manufacturers looking to call their drugs “interchangeable products” must go even further and prove their product can produce the same result as the reference product in any given patient. More information on biologics, biosimilars, and interchangeable products can be found HERE.
Looking Ahead
Medical innovation dates as far back as Neanderthals who we suspect used plants to treat ailments, but has rapidly accelerated over the past 180 years since the first synthetic drug was discovered in the 1860s. The beginnings of the NIH emerged shortly after to examine threats to public health in the 1880s and the FDA emerged in the 1900s to regulate the safety of food and drugs.
Today, both agencies play a critical role in the development of pharmaceutical treatments from conducting basic research to verifying the safety and efficacy of the products in our medicine cabinets. As Congressional leaders continue the appropriations process for fiscal year 2020, expect them to work in a bipartisan fashion to give adequate funding to these agencies, despite the Administration’s objections. Leaders in both parties have long understood the necessary role the public sector plays in partnership with the private sector to bring cures to patients.
Glossary
- Bioequivalence: demonstrates that a generic drug works in the same way and provides the same clinical benefit as the brand name version it is modeled after.
- Biologics: medication created from a variety of natural sources (human, animal, or microorganisms). Biologics are complex to manufacture and are used in the treatment, prevention, or diagnosis of diseases. Biologics are a diverse category of products that can range from vaccines for influenza prevention to gene therapy for cancer treatment.
- Biosimilars: like a generic, a biosimilar is another version of an existing brand name drug; however, they are not the same as generics. Biosimilars have no clinically meaningful differences from existing FDA-approved biologics. Because biological products are so complex to develop, inherent variations caused by the manufacturing process are expected; therefore, a biosimilar must meet the FDA’s high standard of having “no clinically meaningful differences” in purity, safety and effectiveness from the biologic it is being modeled after.
- Brand name drug: refers to a drug marketed under a proprietary, trademark-protected name. “Brand name drugs” or “brands” typically serve as reference products to which generics or biosimilars are compared by the FDA.
- Generic: medication deemed by the FDA to be the same as already marketed brand-name drugs in dosage form, safety, strength, and other factors. These similarities demonstrate bioequivalence, or prove that a generic medicine works in the same way and provides the same clinical benefit as the brand version.
- Reference product: brand name drug product, already approved by FDA, against which a proposed generic or biosimilar product is compared for purposes of safety and efficacy.
A full glossary of common health care terms, including several used in this Basic, can be found HERE.
Additional Resources
- CDC – Therapeutic Drug Use
- CNBC – FDA Commissioner Scott Gottlieb
- FDA – Biosimilars
- FDA – Biosimilar and Interchangeable Products
- FDA – Biosimilar Development, Review, and Approval
- FDA – Drug Development Process
- FDA – Generic Drugs
- FDA – Generic Drug Approval Process
- MedPAC – Drug Development and Supply Chain
- NEJM – Role of Public-Sector Research
- PNAS – Contribution of NIH funding
- World Health Organization – Clinical Trials